|
Was sind die Liganden von PPARg?
Nach einem physiologisch relevantem Liganden, der mit hoher Affinität an
PPARg bindet, wird immer noch gefahndet. Zwar sind einige natürlicherweise
vorkommende Aktivatoren von PPARg gefunden worden, doch solche
Fettsäure-Derivate wie 15-Deoxyprostaglandin J2,
9-Hydroxyoktadekadien-Säure (9-HODE), 13-HODE und Linolensäure
sind erst in relativ hoher Konzentration wirksam.
Die bekanntesten synthetischen Verbindungen, die als Liganden von PPARg
fungieren, sind verschiedene Thiazolidindione (TZD). Diese oralen Antidiabetika
wirken über PPARg einer Insulin-Resistenz entgegen.
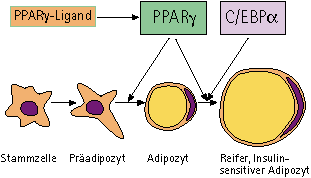
PPARg: Essentieller Faktor bei der Adipogenese
Fettzellen können sich ein Leben lang aus fibroblastartigen Vorläuferzellen
(Adipoblasten) des Fettgewebes neu bilden. Nach Stimulierung durch Wachstumsfaktoren
proliferieren Adipoblasten und differenzieren sich bei Einwirkung verschiedener
Faktoren — von denen PPARg essentiell ist — zu reifen
Insulin-sensitiven Adipozyten aus.
PPARgamma ist zwar ein essentieller aber nicht der einzige Transkriptionsfaktor,
über dessen Aktivität das Heranreifen neuer Fettzellen gesteuert wird. Zur
vollen Ausdifferenzierung von Adipozyten tragen noch zwei weitere Transkriptionsfaktoren
bei. Deren Bezeichnungen sind ähnlich verwirrend wie die von PPARg: Ihre
kompliziert anmutenden Kürzel lauten ADD-1/SREBP-1 und C/EBP. Diese
Transkriptionsfaktoren bewirken unter anderem, daß Adipozyten spezifische
Proteine herstellen, die sich bei Präadipozyten noch nicht nachweisen lassen,
d.h. sie prägen den Phänotyp der ausgereiften Fettzellen. Darüber
hinaus beeinflussen sich die fettgeweblichen Transkriptionsfaktoren auch gegenseitig.
Wird z.B. ADD-1/SREBP-1 aktiviert, führt das zu vermehrter Bildung von PPARg.
Und damit letzterer voll wirksam werden kann, wird über Umwege auch gleich die
Bildung von PPARg-Liganden mit angeregt (2).
Welche Rolle spielt PPARg in ausgereiften Adipozyten?
Anhand von Tiermodellen konnte nachgewiesen werden, daß die Menge an
PPARg in den Adipozyten bei fettreicher Ernährung einen entscheidenden
Faktor für hypertrophes Zellwachstum und die Entwicklung von Insulinresistenz
darstellt. In diesbezüglichen Experimenten wurden Mäuse mit nur einem
intakten Allel des PPARg-Gens untersucht. Solche
PPARg-heterozygoten
Individuen produzieren signifikant weniger PPARg-Moleküle als Wildtyp-Mäuse,
bei denen beide entsprechenden Allele intakt sind.
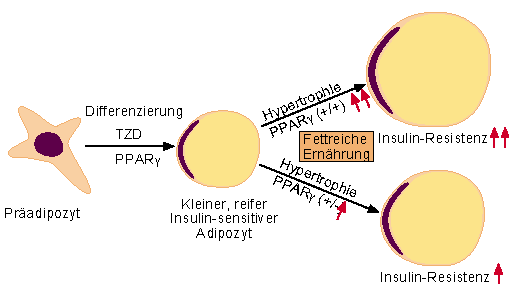
Abb. 2: PPARg fungiert als Vermittler von
Adipozyten-Hypertrophie und Insulin-Resistenz: Sind beide Kopien des
PPARg intakt (PPARg +/+), ist die Bildung riesiger, stark die
Insulin-Resistenz fördernder Adipozyten begünstigt. Bei
Heterozygoten ist ein Allel defekt (PPARg +/-), so daß geringere
Mengen an PPARg-Protein exprimiert wird. Das schützt bis zu einem
gewissen Grad vor Adipositas und Insulin-Resistenz. Die Wirkung sehr
starker Liganden von PPARg (Thiazolidindione TZD) führt zur Bildung
zahlreicher kleiner, Insulin-sensitiber Adipozyten (nach Kubota et al, 1999).
Bei normaler Verköstigung erleiden die Gen-defekten Mäuse keine
gesundheitlichen Schäden, und erstaunlicherweise kann ihnen auch eine fettreiche
Ernährung nicht viel anhaben: Die Adipozyten bleiben relativ klein, und die
Insulin-Resistenz ist nur geringgradig ausgeprägt. Hingegen wachsen
die Adipozyten bei Wildtyp-Mäusen unter den gleichen Ernährungsbedingungen
zu Riesenzellen heran und lösen über Faktoren wie den
(TNF-a) und freie Fettsäuren eine gravierende Insulin-Resistenz aus (4).
Wenn Nahrung intermittierend knapp ist, sorgt PPARg offenbar dafür,
daß ein Teil der in guten Zeiten verfügbaren Energie zur
Überbrückung von Mangelzuständen aufgespart wird. In dieser
Beziehung unterscheiden sich Menschen kaum von Mäusen. Denn nur durch
eine Strategie der Vorratshaltung ist ein Überleben von Hungerperioden
überhaupt möglich. Doch in Zeiten anhaltenden Nahrungsüberangebots
kehrt sich dieser haushälterische Umgang mit Energieträgern ins Gegenteil,
und Adipositas wird zur »Volksseuche«.
Waren über Äonen evolutionärer Entwicklung zwei intakte Allele
des PPARg-Gens den begrenzten alimentären Ressourcen ideal angepaßt,
wären wir heute offenbar besser dran, könnten wir — wie bei
Labormäusen — eines davon ausschalten. Aber auch wenn das Utopie
ist, ergeben sich aus dieser Erkenntnis Ansätze für die Entwicklung
pharmazeutischer Präparate wie beispielsweise von PPARg-Antagonisten.
Besserung einer Insulin-Resistenz über PPARg
Zu den therapeutisch als Anti-Diabetika genutzten Liganden von PPARg
gehören verschiedene Thiazolidindione (TZD), die einer Insulinresistenz
entgegenwirken. Das mag im Lichte der vorangegangenen Schilderung, wonach
fettreiche Ernährung besser vertragen wird, wenn die PPARg-Aktivität
gedrosselt ist, als Paradox erscheinen. Es stellte sich jedoch heraus, daß
über starke Liganden wie TZD insbesondere die Anzahl kleiner,
Insulin-sensitiven Adipozyten signifikant vermehrt und zugleich die Anzahl
der großen Adipozyten, die TNF-a produzieren und Insulinresistenz
fördern, vermindert wird.
1. Kubota N, Terauchi Y, Miki H, et al.1999. PPARg
mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 4:597-609.
2. Kim JB, Wright HM, Wright M, Spiegelman BM. 1998. ADD1/SREBP1 activates
PPARg through the production of endogenous ligand.
Proc Natl Acad Sci USA 95:4333-4337.
3. Brun RP, Spiegelman BM. 1997. PPARg and the
molecular control of adipogenesis. J Endocrinol 155:217-218.
4. Rosen ED, Sarraf P, Troy AE, et al. 1999. PPARg
is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4:611-617.
5. Okuno A, Tamemoto H, Tobe K, et al. 1998. Triglitazone increases the
number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats.
J Clin Invest 101:1354-1361.
6. Deeb SS; Fajas L; Nemoto M, et al. 1998. A Pro12Ala substitution in
PPARg2 associated with decreased receptor activity, lower body mass index
and improved insulin sensitivity. Nat Genet 20:284-287.
Reviews
1. Desvergne B, Michalik L, Wahli W. 2004. Be fit or sick: peroxisome
proliferator-activated receptors are down the road. Mol Endocrinol 18:1321-1332.
2. Ferré P. 2004. The biology of peroxisome proliferator-activated receptors. Diabetes
53(suppl. 1):S43-S50.
3. Auwerx J. 1999. PPARg, the ultimate
thrifty gene. Diabetologia 42:1033-1049.
4. Lowell BB. 1999. PPARg: An essential regulator
of adipogenesis and modulator of fat cell function. Cell 99:239-242.
5. Brun RP, Kim JB, Hu E, Spiegelman BM. 1997. Peroxisome proliferator-activated
receptor gamma and the control of adipogenesis. Curr Opin Lipidol 8:212-218.
|